Using the Taxonomic Name Standardization App
taxonomic-app.RmdIntroduction
The launch_taxonomic_match_app() function provides an
interactive Shiny application for standardizing taxonomic names against
the Central African plant taxonomic backbone database. This visual
interface is ideal for:
- Exploring and cleaning taxonomic data interactively
- Understanding match quality through visual feedback
- Manually reviewing uncertain matches
- Enriching data with species-level traits from the database
- Checking taxonomic name provenance via WCVP integration
Prerequisites
With database credentials (full access)
To access all features including traits enrichment, you need database
credentials configured (see setup_db_credentials()). Once
launched, the app presents a login screen where you enter your
credentials.
Without credentials (public access mode)
Since March 2026, the app can be launched without any database credentials. Click “Use offline (cached backbone)” on the login screen to enter offline mode:
- Automatic matching and fuzzy suggestions work via a cached local backbone
- Manual review is fully functional
- Traits enrichment is hidden (requires a live database connection)
- A “Read-only” badge is displayed to indicate limited permissions
This is useful for exploring the matching workflow, standardizing names without a personal account, or working from locations without network access to the database.
Quick Start
Launch the app with a single command:
Alternatively, pre-load your data or set options:
# With R data.frame
my_data <- read.csv("tree_inventory.csv")
launch_taxonomic_match_app(data = my_data, name_column = "species_name")
# Launch in English (default is French)
launch_taxonomic_match_app(language = "en")
# Adjust fuzzy matching sensitivity (default is 0.7)
launch_taxonomic_match_app(min_similarity = 0.5) # More permissive matchingStep-by-Step Walkthrough
Phase 1: Initial View
When you first launch the app, you see a login screen. After authenticating (or choosing offline mode), the main interface appears with a sidebar for configuration and tabs for different workflow phases:
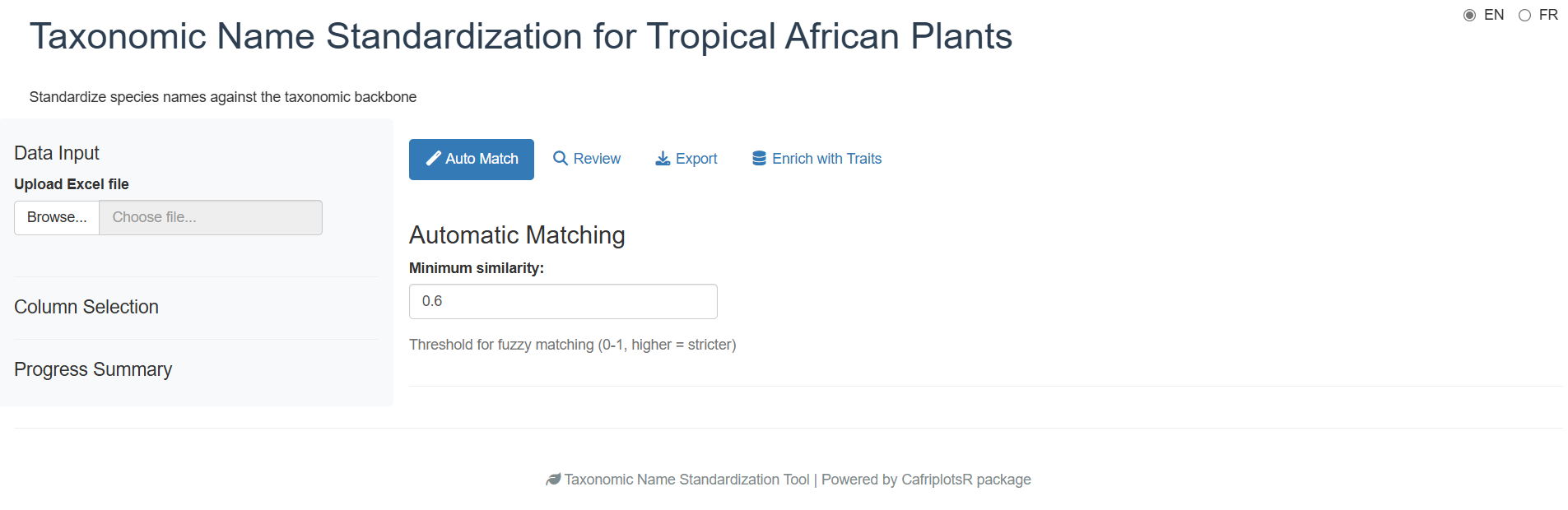
The app uses a tabbed workflow that guides you through each phase sequentially:
- Auto Match — Automatic matching
- Review — Manual review of unmatched names
- Export — Download results
- Traits Enrichment — Add species traits (hidden in offline mode)
Phase 2: Upload Your Data
The first step is to provide your data. The app offers two input methods:
File Upload (Default)
- Upload an Excel file using the file browser (supports .xlsx, .xls); for multi-sheet files you can select which sheet to use
- Upload a CSV file
-
Use pre-loaded R data (if you passed the
dataparameter)
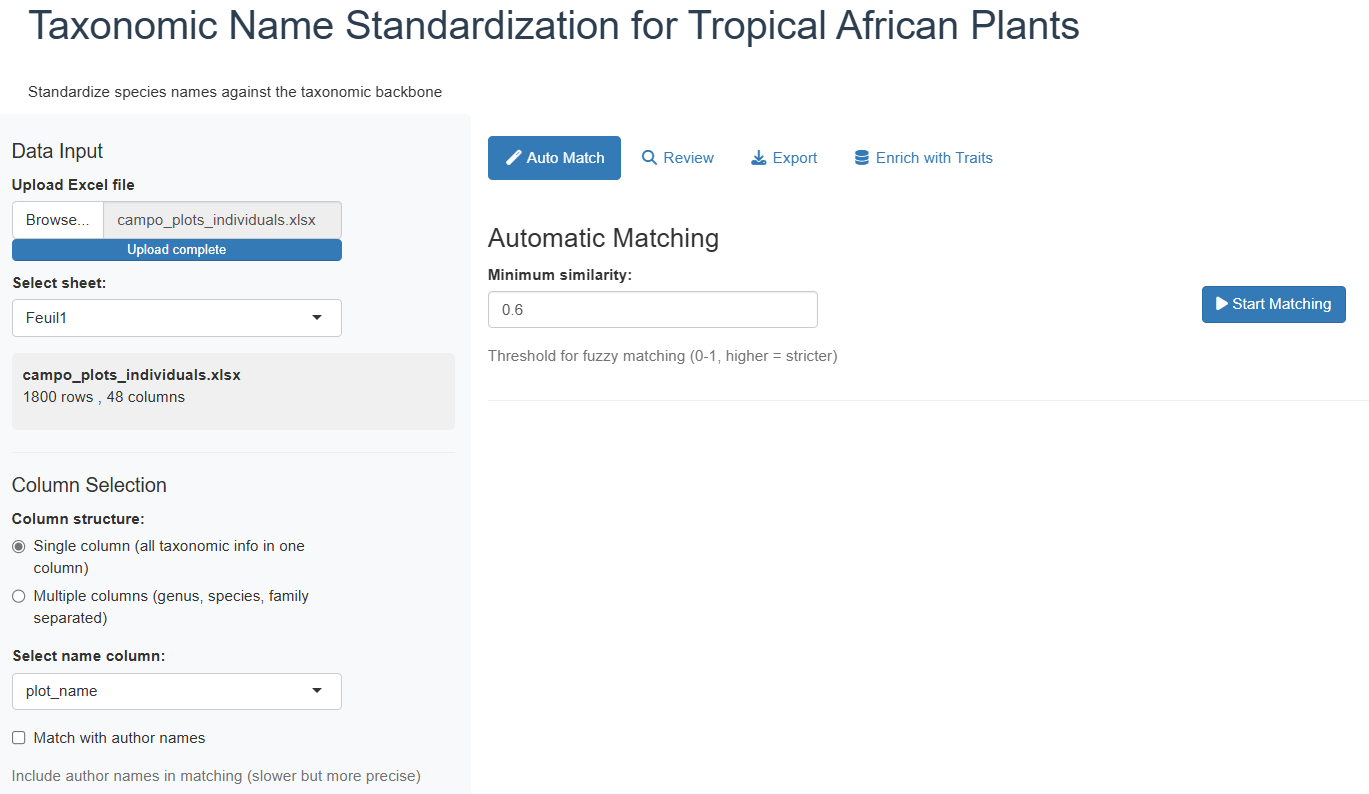
The app displays a preview of your uploaded data so you can verify it
was read correctly. Excel files are read with
guess_max = 30000 to improve column type detection for
large files.
Text Input (Copy-Paste)
For quick standardization of a few names, or when you have a list copied from another source, use the Text input method:
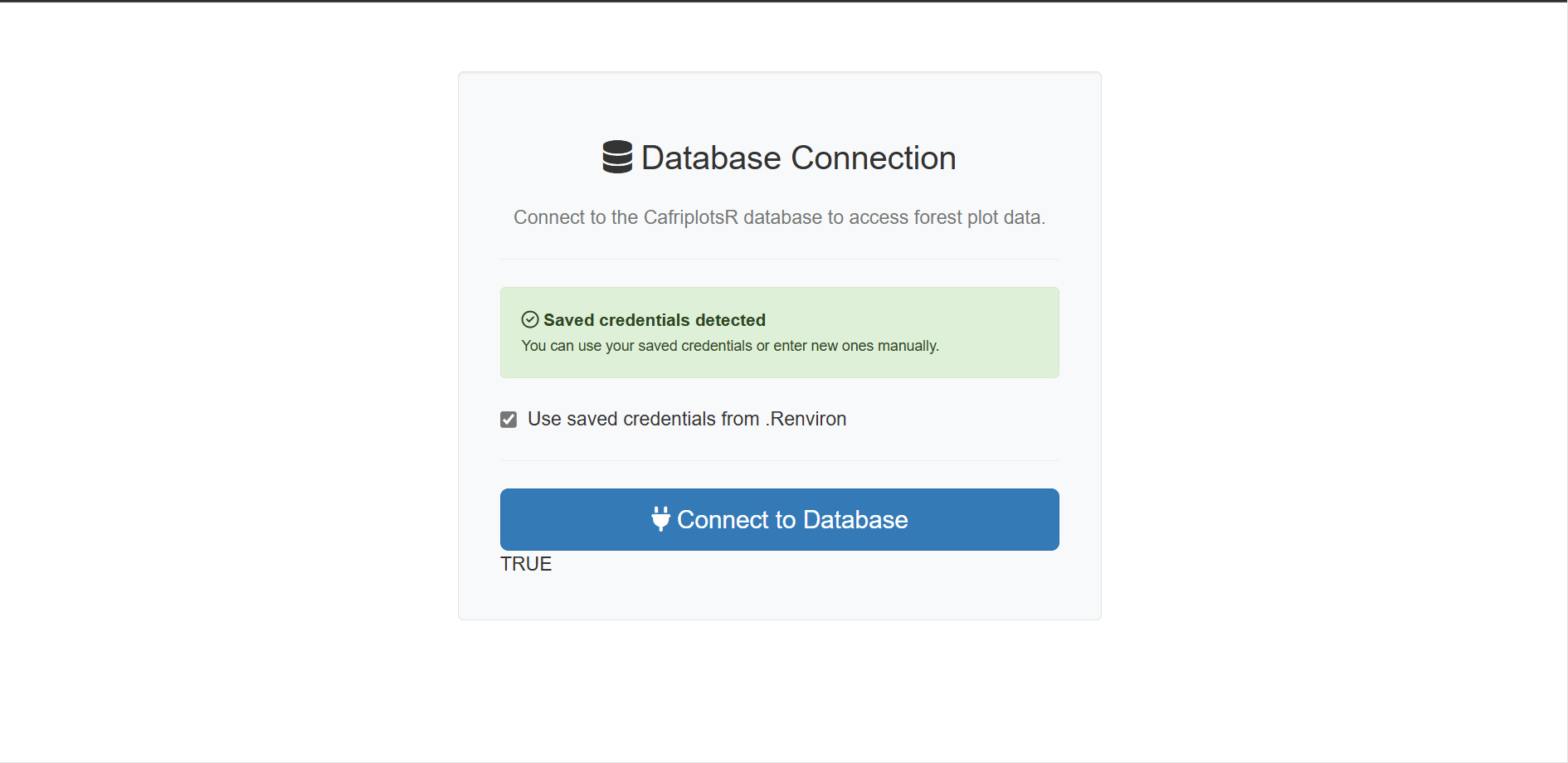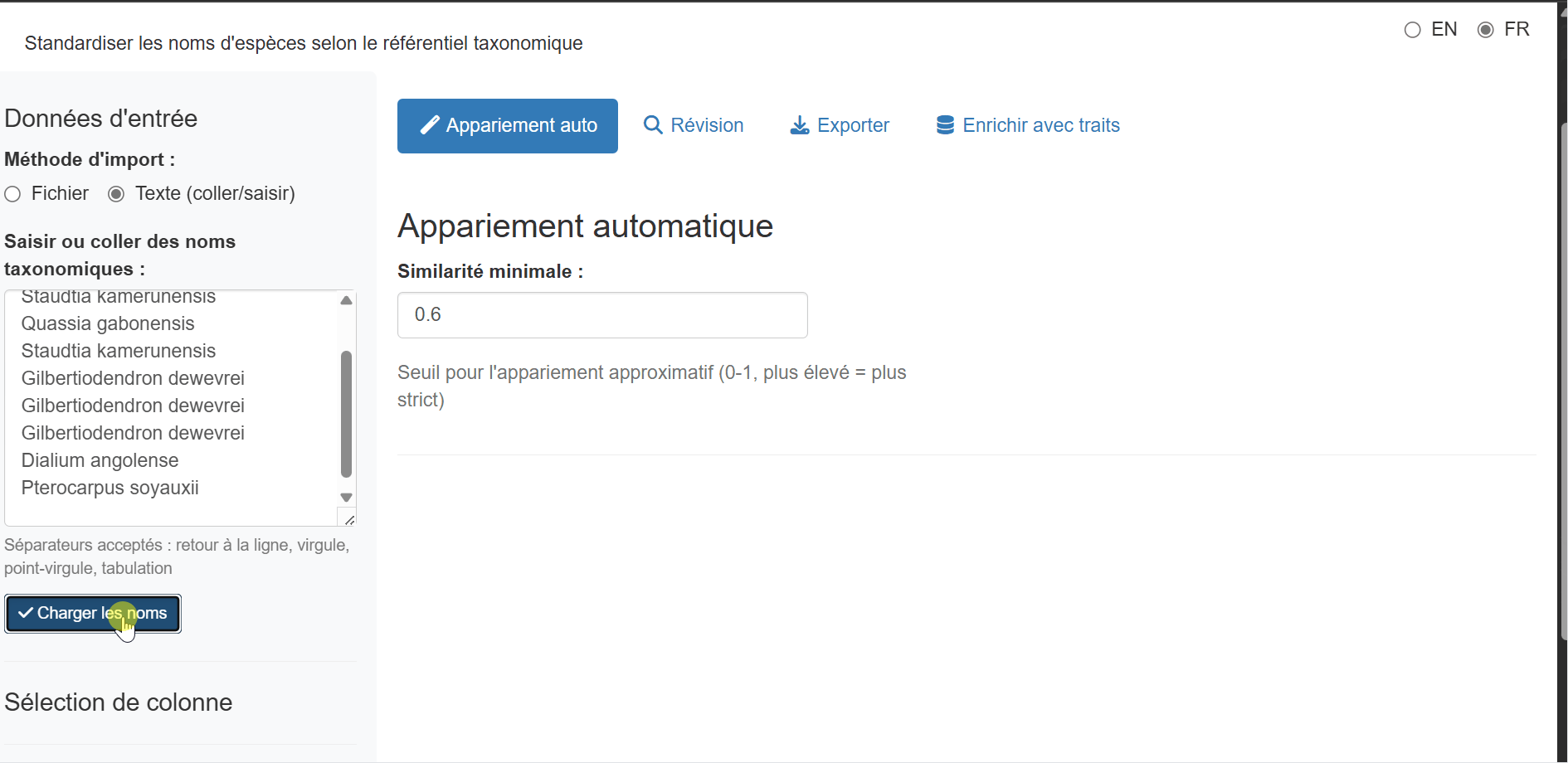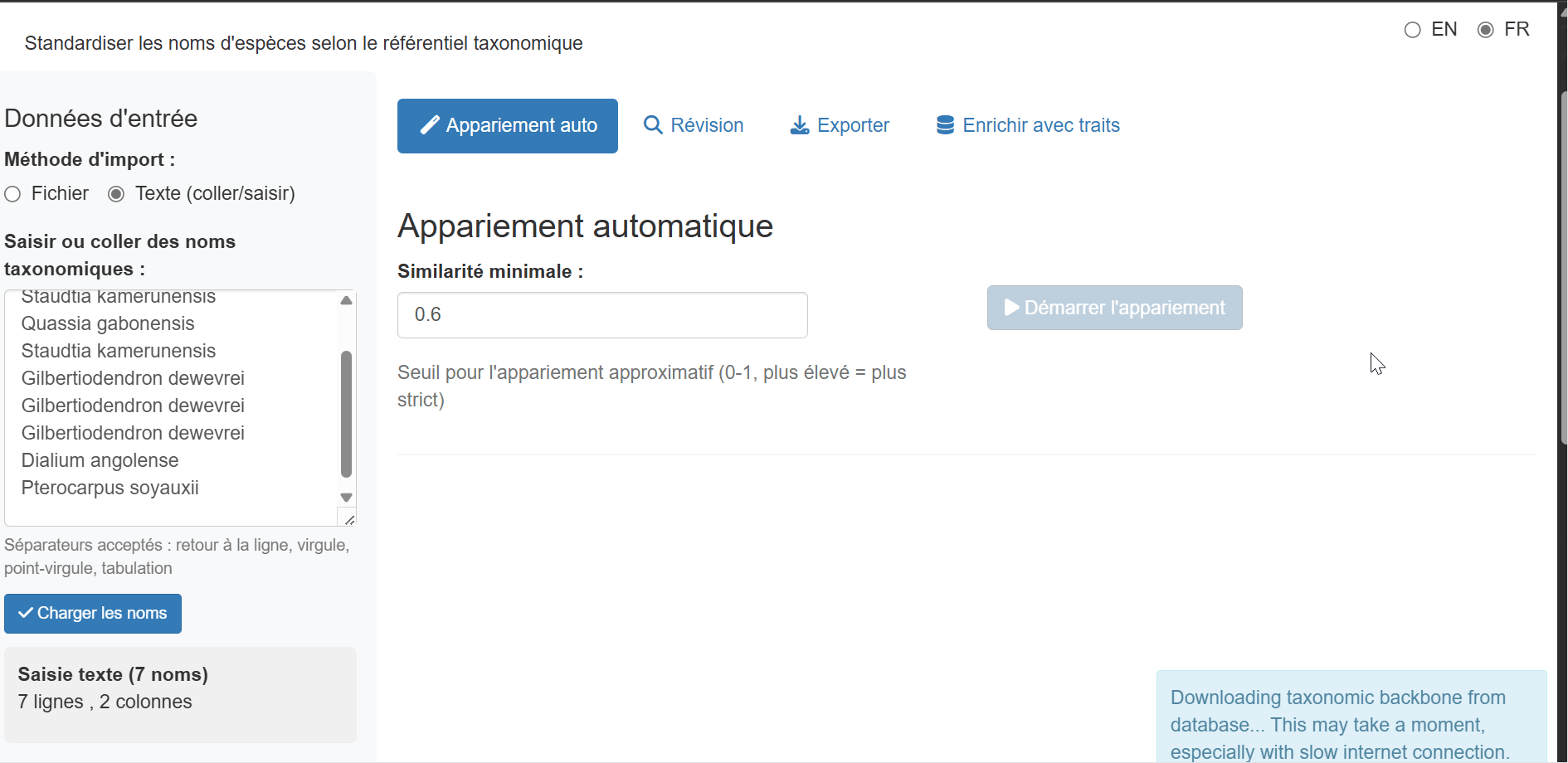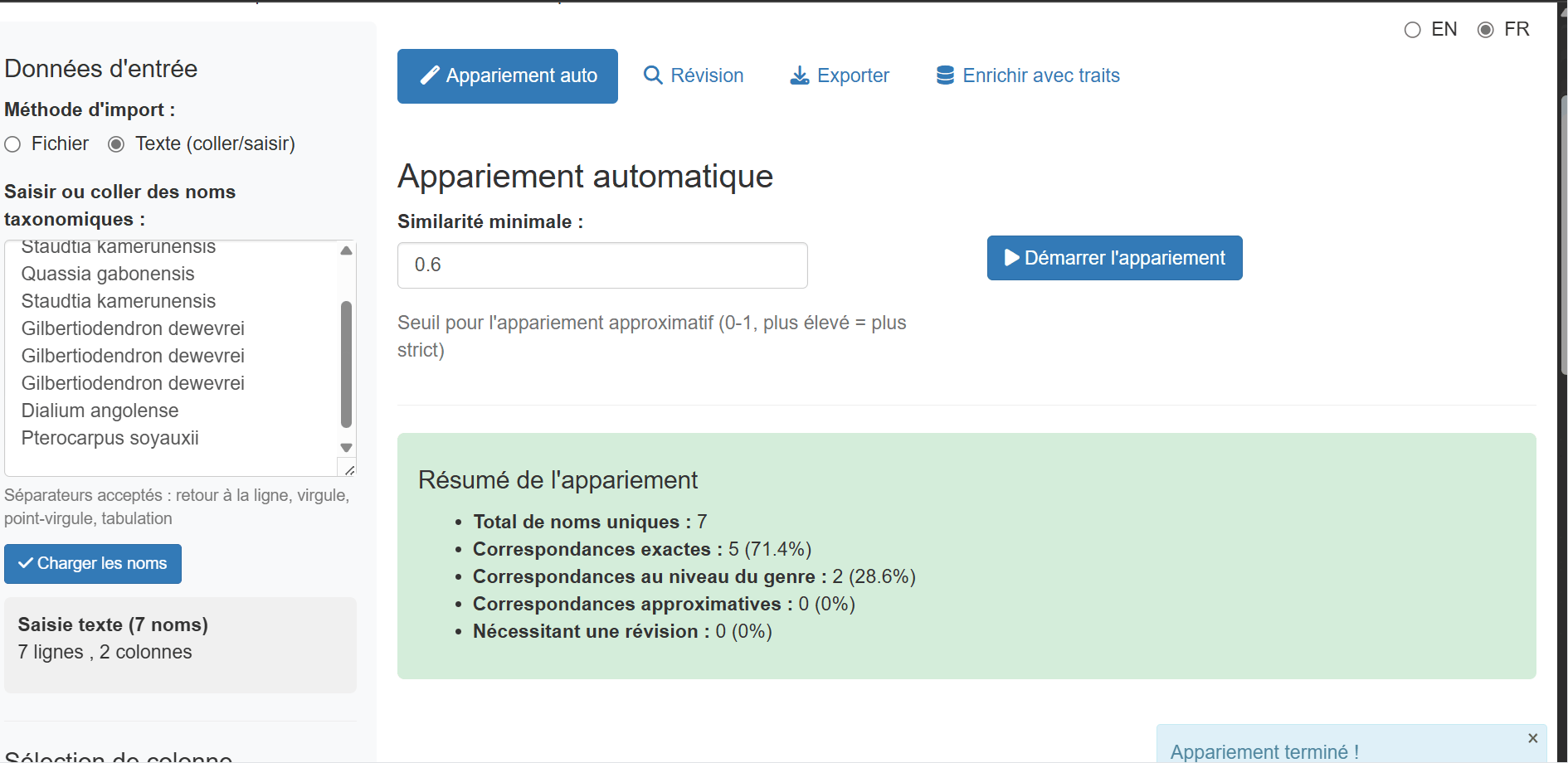
- Select “Text input (paste/type)” from the input method radio buttons
- Paste or type your taxonomic names in the text area
- Click “Load names” to process the input
Accepted separators: - One name per line
(recommended) - Comma-separated:
Lophira alata, Terminalia superba, Aucoumea klaineana -
Semicolon-separated:
Lophira alata; Terminalia superba; Aucoumea klaineana -
Tab-separated (useful when pasting from Excel)
The app automatically removes empty lines, trims whitespace, and
deduplicates names while preserving order. A single column named
taxon_name is created for matching.
Phase 3: Select Name Column(s)
Once data is loaded, you have two options for selecting taxonomic names:
Single Column Mode (Default)
Select one column containing the full taxonomic name:
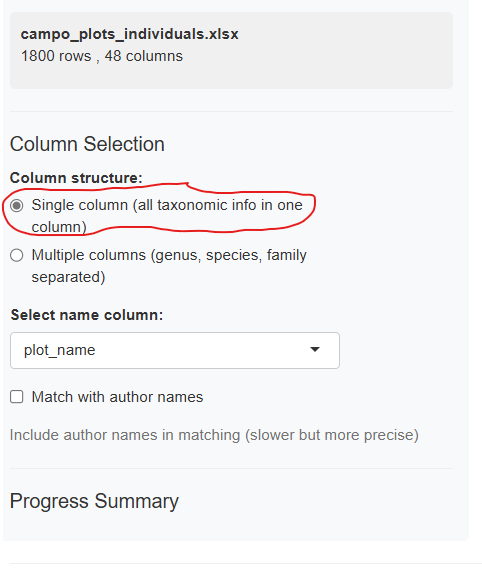
The dropdown menu shows all available columns from your dataset. Choose the one containing species names (typically formatted as “Genus species” or “Genus species Author”).
Multiple Column Mode
If your data has separate columns for genus, species, and family, enable “Use multiple columns”:
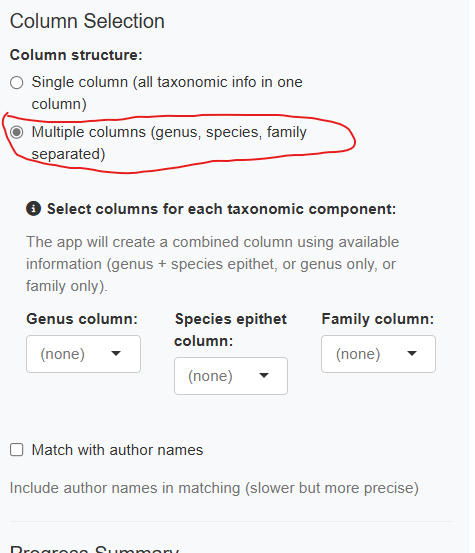
The app combines these columns hierarchically: - Genus + species available → “Genus species” - Genus only → “Genus” - Family only → “Family”
You can also optionally include an author column.
Phase 4: Automatic Matching
Click the “Start Matching” button to begin the automatic matching process. The app uses a five-tier matching strategy:
- Exact match on species: Direct lookup of full name (genus + species)
- Exact match on genus: Match at genus level
- Exact match on family: Match at family level
- Exact match on class: Match at class level (e.g., names ending in -opsida, -psida)
-
Fuzzy matching: Approximate string matching
(trigram-Jaccard via
stringdist) for remaining names
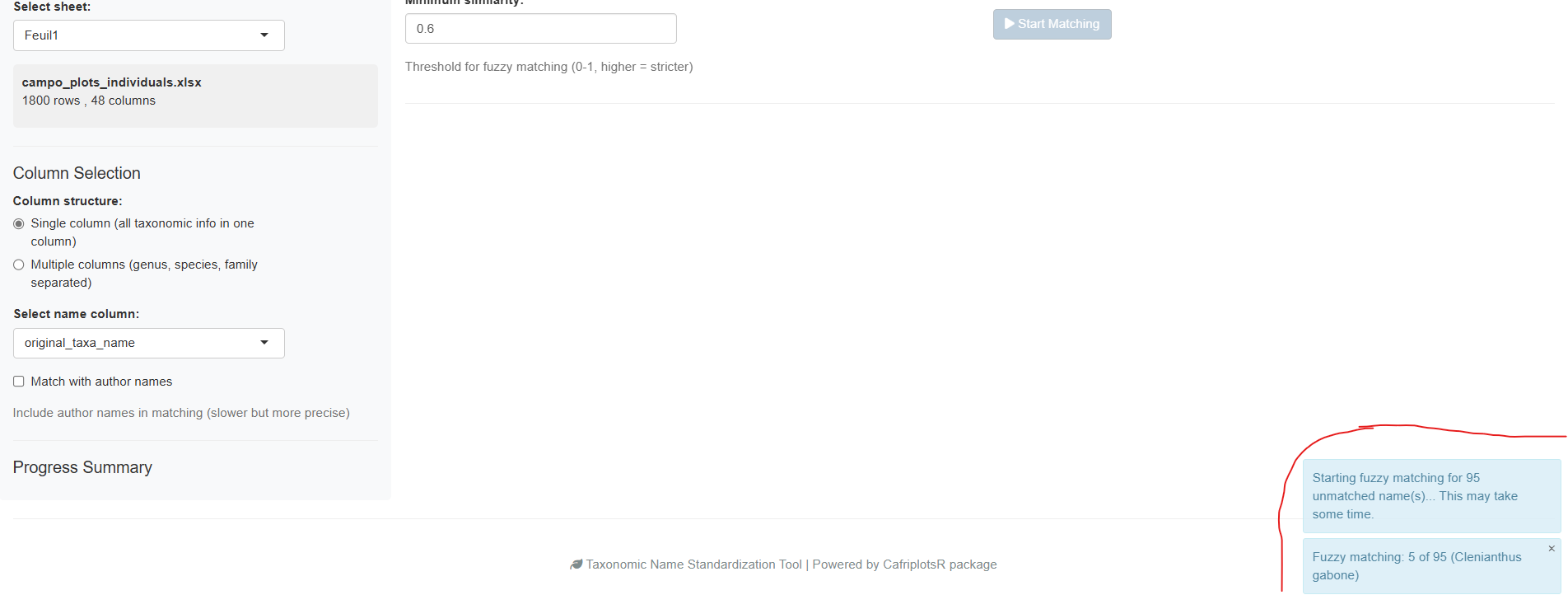
The progress bar shows real-time status and correctly accounts for manually reviewed names in the completion percentage. The sidebar displays live statistics:
- Number of exact matches
- Number of genus-level matches
- Number of fuzzy matches
- Number of unmatched names
Checkpoint / resume: Matching progress is automatically saved to a temporary file. If you accidentally close the browser tab, re-opening the app will offer to resume from where you left off.
Phase 5: Review Match Results
After matching completes, the Auto Match tab shows a summary table with all names and their match status:
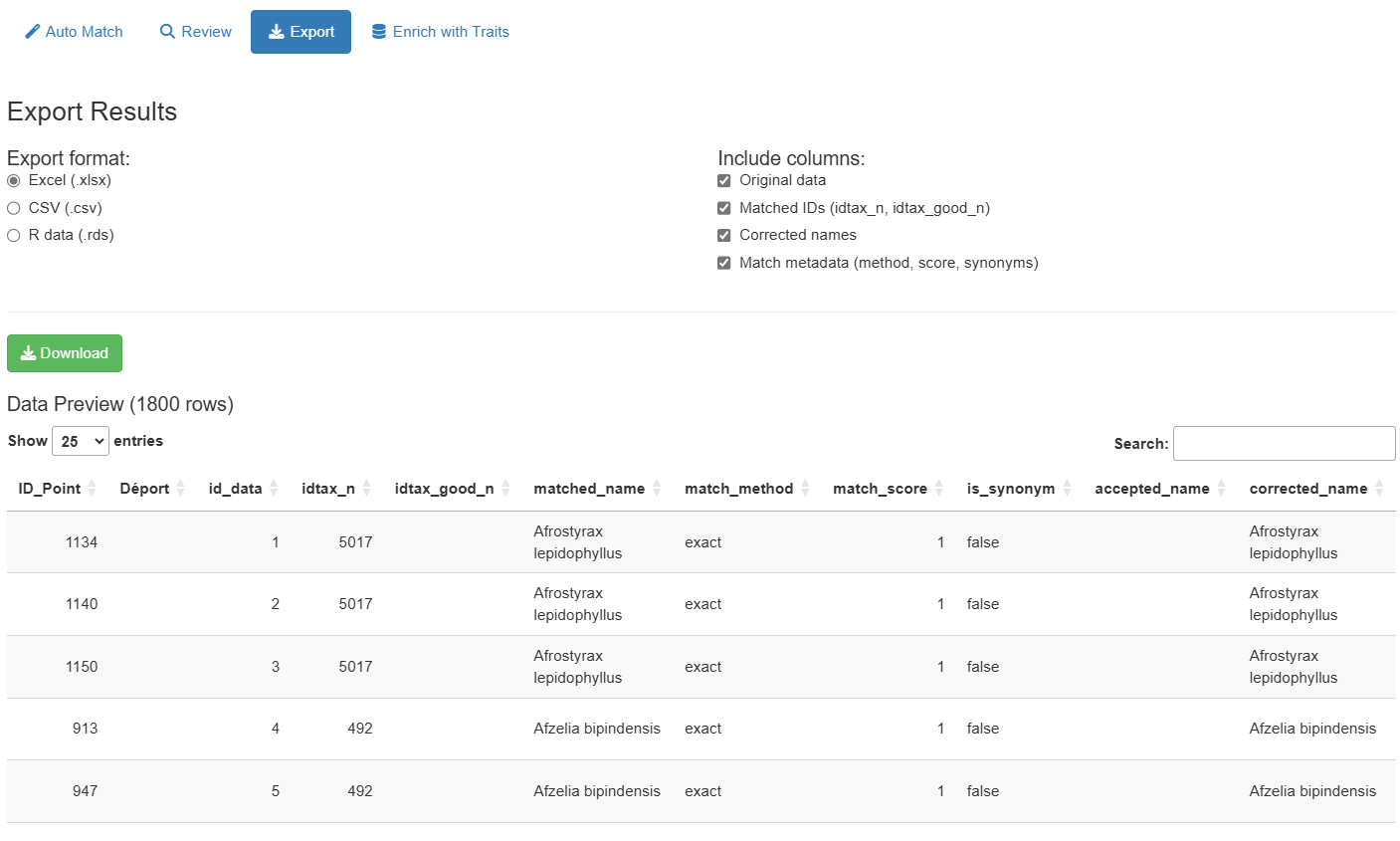
The results table includes:
- Original name: Your input name
- matched_name: Name found in backbone
-
match_method: How it was matched
(
exact_species,exact_genus,exact_family,exact_class,fuzzy,manual) - match_score: Similarity score (0–1, higher is better)
- idtax_n: Taxon ID in database
- is_synonym: Whether matched name is a synonym
- accepted_name: Current accepted name (if synonym)
Match quality indicators:
- Exact match (1.0): Perfect match, no review needed
- High similarity (>0.8): Very likely correct, quick review recommended
- Medium similarity (0.5–0.8): Possible match, review suggested
- Low similarity (<0.5): Uncertain, manual review required
- No match: Requires manual selection
Phase 6: Manual Review
For unmatched or uncertain names, switch to the “Review” tab to manually review and select matches:
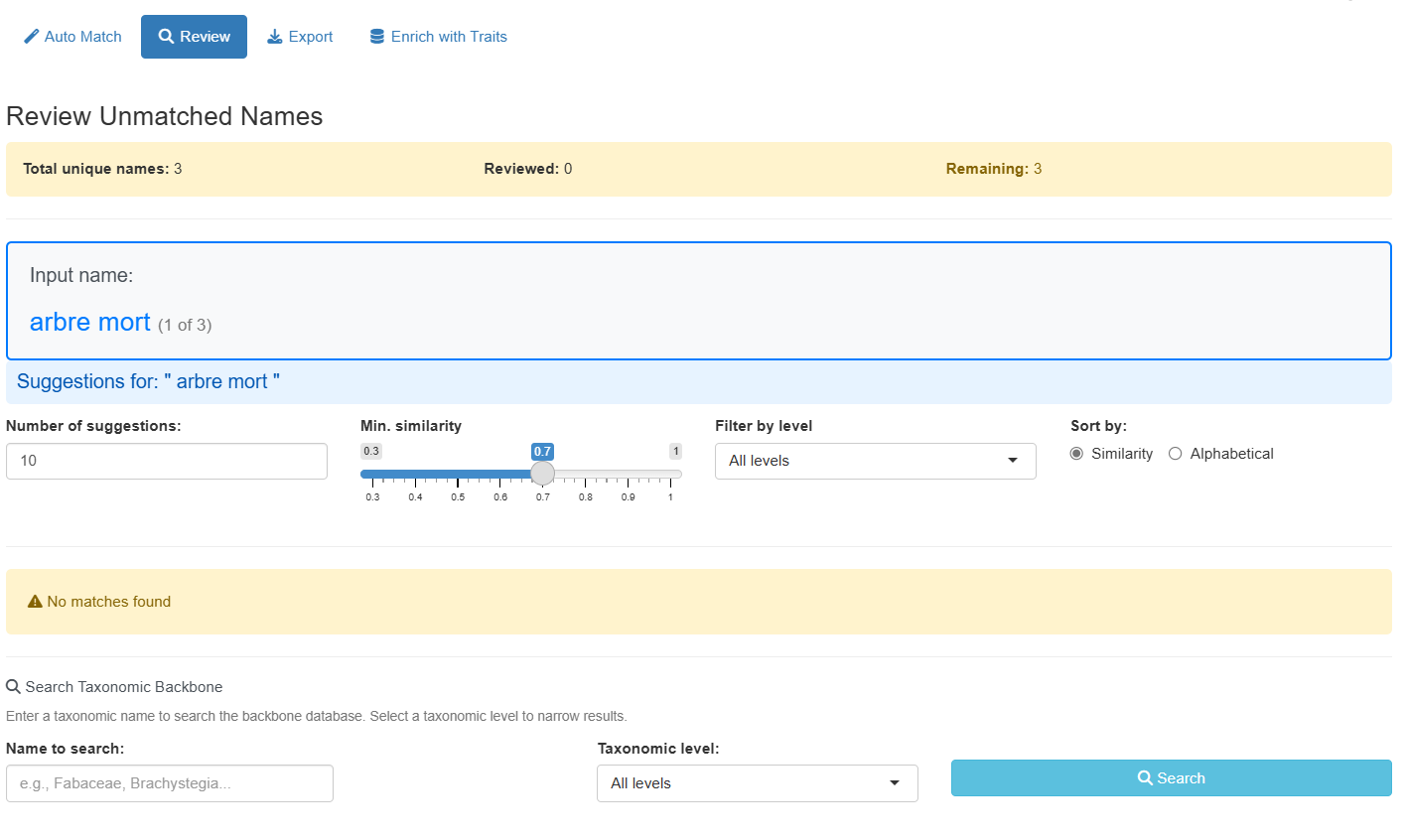
The review interface provides two ways to find matches:
Fuzzy Suggestions Panel
Shows automatic suggestions ranked by similarity with advanced filtering options:
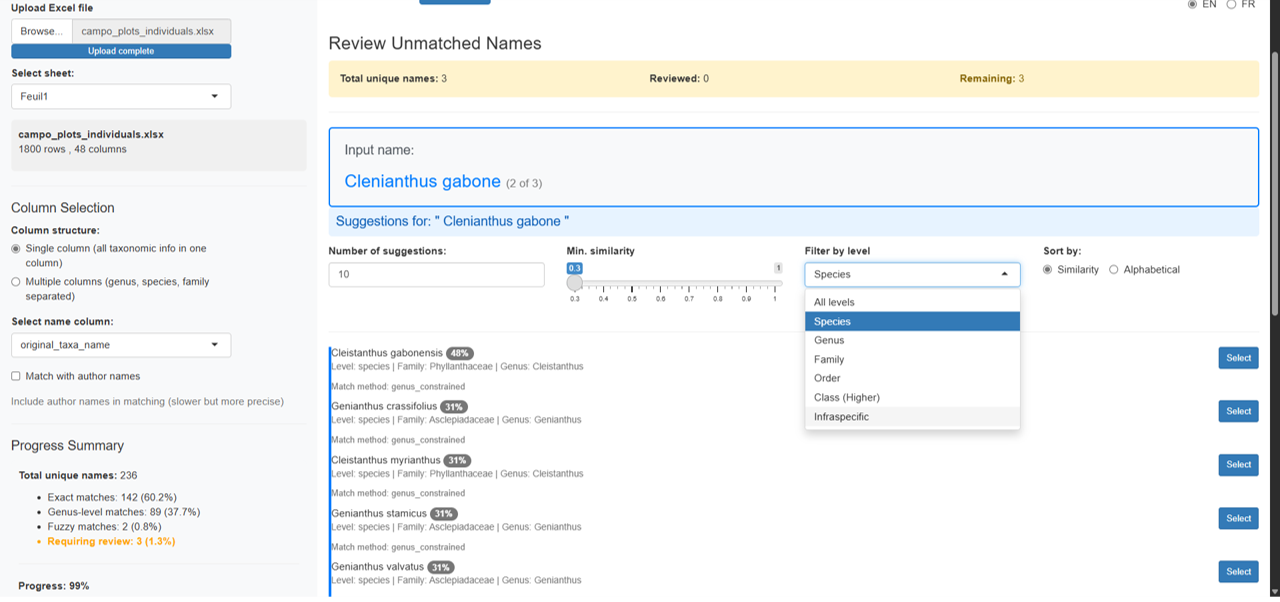
Filtering options:
- Number of suggestions: Slider to show 5–30 suggestions
- Minimum similarity: Adjust threshold (0.3–1.0)
- Taxonomic level filter: Filter by All, Species, Genus, Family, Order, Class, or Infraspecific
- Sort by: Similarity score or alphabetical order
Each suggestion card displays:
- Name with color-coded similarity badge (green = high, blue = medium, yellow = low)
- Taxonomic level and family
- Synonym information if applicable
- Select button for one-click acceptance
Manual Search Panel
For names without good suggestions, use the manual search:
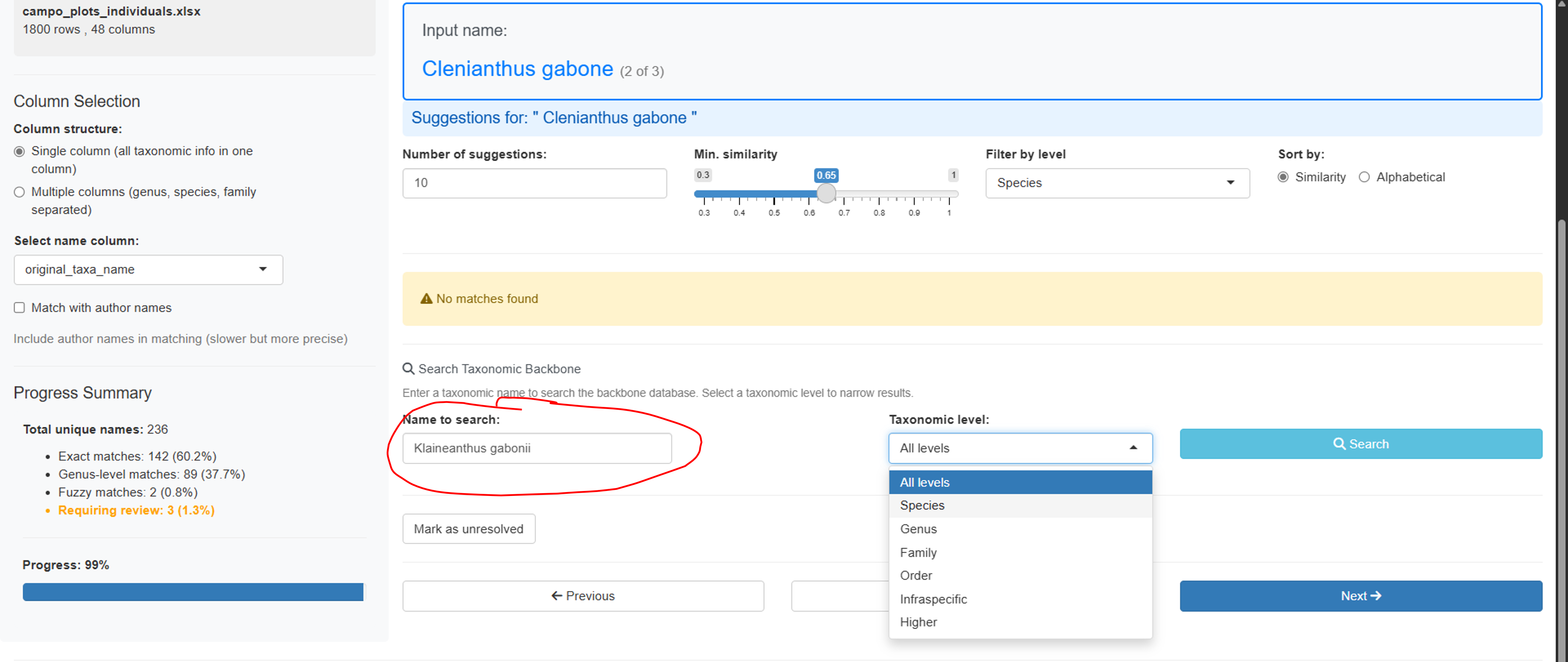
- Type any search term to query the taxonomic backbone
- Filter results by taxonomic level
- View detailed information for each match
- Select the correct match or mark as “unresolved”
Navigation:
- Use Previous/Skip/Next buttons to browse unmatched names
- Progress counter shows reviewed vs. remaining names
- The app remembers your selections and automatically updates the results
Phase 7: Enrich Data with Traits
Switch to the “Traits Enrichment” tab to add species-level traits to your matched data (requires a database connection; this tab is hidden in offline mode):
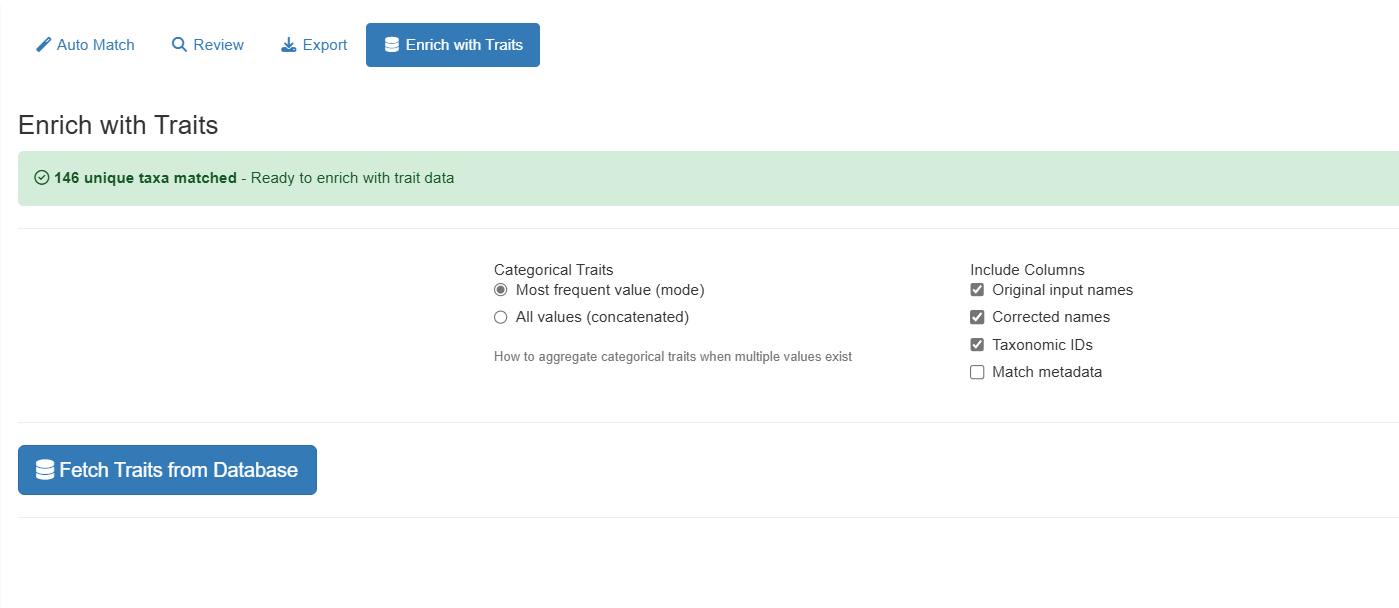
Options:
-
Categorical aggregation mode:
- “mode” — Use most frequent value per taxon
- “concat” — Concatenate all unique values
-
Select columns to include:
- Original input names
- Corrected names
- Taxonomic IDs
- Match metadata
Available traits include growth form, wood density, leaf traits, and ecological characteristics.
The enriched data combines your matched taxa with selected traits. A wide format (one row per taxon, traits as columns) and a long format (one row per taxon × trait combination) are both available as separate sub-tabs:
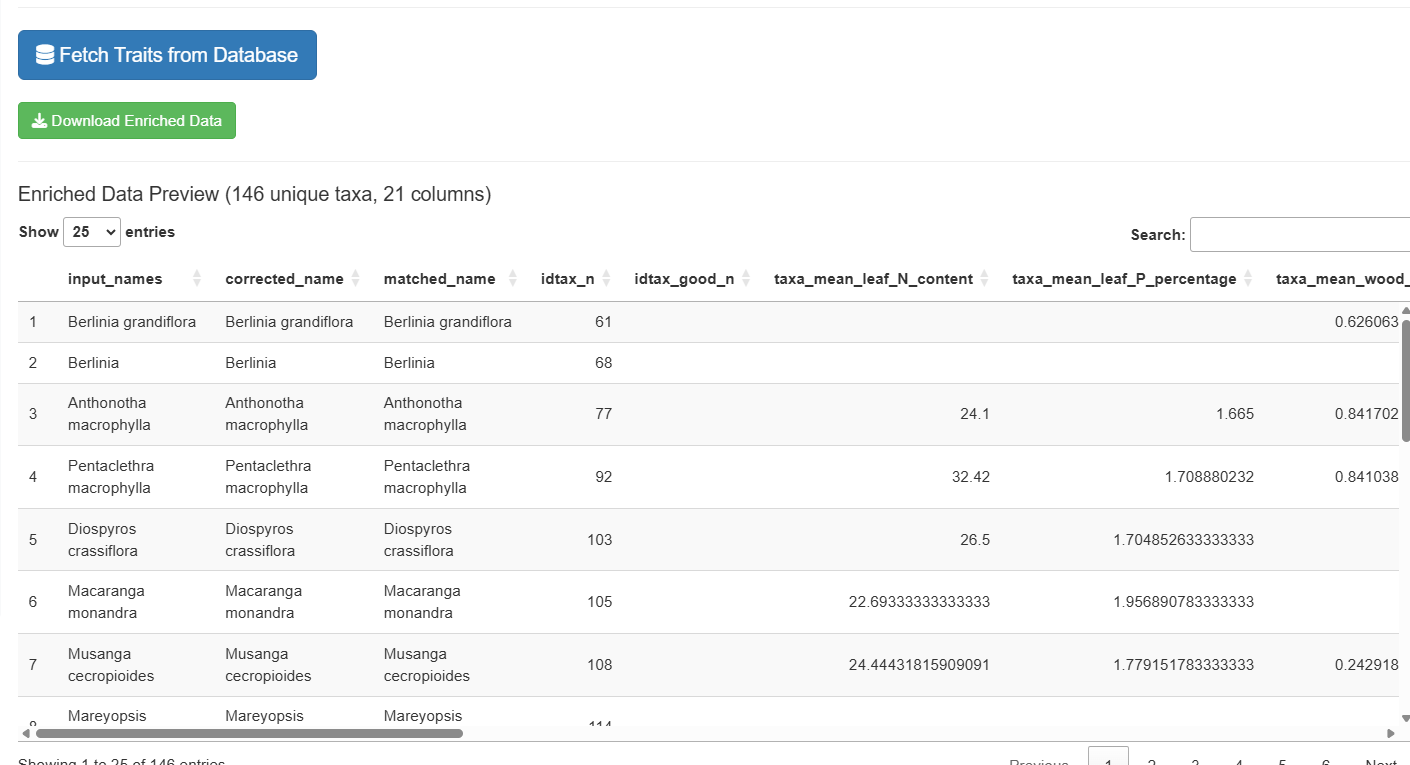
Note: The enriched export creates one row per unique taxon, not per input row. Input names are concatenated with pipe separators.
Phase 8: Export Results
Switch to the “Export” tab to download your standardized dataset:
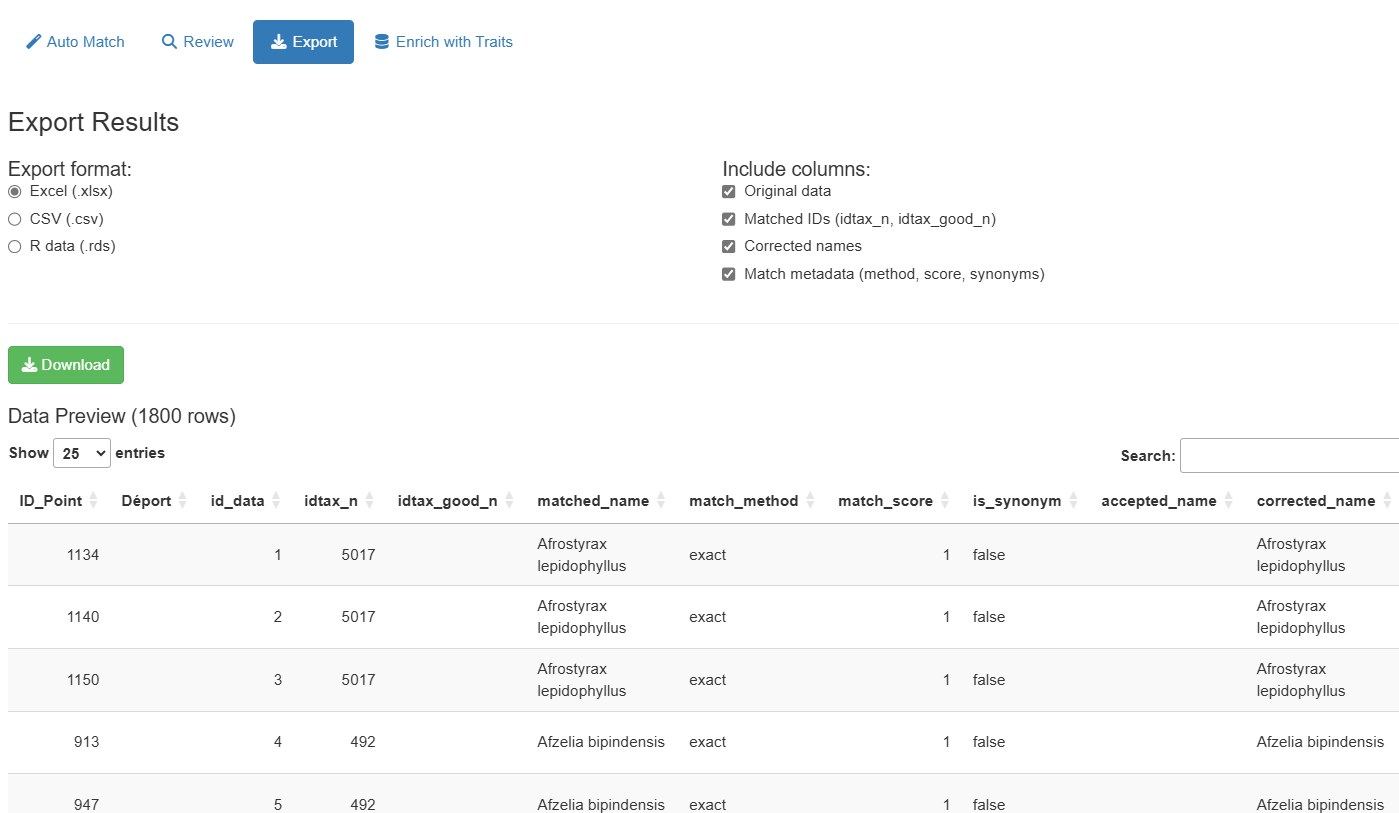
Available formats:
- Excel (.xlsx): Best for sharing with collaborators
- CSV (.csv): Universal tabular format
- RDS (.rds): R-native format preserving data types
Selectable columns:
- Original data (all your input columns)
- Matched IDs (
idtax_n,idtax_good_n) - Corrected names (
corrected_name,matched_name) - Match metadata (
match_method,match_score,is_synonym,accepted_name) - WCVP columns (
wcvp_plant_name_id,wcvp_accepted_plant_name_id) — see WCVP section below
A preview table shows the data before export with pagination controls.
Understanding Output Columns
The app adds these columns to your data:
| Column | Description |
|---|---|
idtax_n |
Matched taxon ID in backbone database |
idtax_good_n |
Accepted taxon ID (for synonyms) |
matched_name |
Name found in backbone |
corrected_name |
Final standardized name |
name_source |
Which backbone was used for the match (internal, WCVP, etc.) |
match_method |
Matching strategy used (exact_species,
exact_genus, exact_family,
exact_class, fuzzy, manual,
unresolved) |
match_score |
Similarity score (0–1) |
is_synonym |
TRUE if matched name is a synonym |
accepted_name |
Current accepted name (if synonym) |
family |
Taxonomic family |
genus |
Taxonomic genus |
wcvp_plant_name_id |
WCVP ID for the matched name (optional, see below) |
wcvp_accepted_plant_name_id |
WCVP ID for the accepted name (optional, see below) |
Advanced Options
Language Selection
The app supports bilingual operation with French and English interfaces. French is the default language.
A language toggle is located in the top-right corner of the app: - Click “FR” for French interface - Click “EN” for English interface
The switch is instant and affects all UI elements. To set the initial language programmatically:
# Launch app in English
launch_taxonomic_match_app(language = "en")
# Launch app in French (default)
launch_taxonomic_match_app(language = "fr")WCVP Integration
The app can optionally enrich results with World Checklist of Vascular Plants (WCVP) identifiers. When the taxa database contains WCVP data, a “Use WCVP names in output” checkbox appears in the sidebar.
Enabling it adds two columns to your export:
-
wcvp_plant_name_id— WCVP identifier for the matched name -
wcvp_accepted_plant_name_id— WCVP identifier for the accepted name
The name_source column records which backbone was used,
helping you document the taxonomic provenance of each match.
Adjusting Fuzzy Matching
Control matching sensitivity with the min_similarity
parameter:
# Very strict - only high-quality matches
launch_taxonomic_match_app(min_similarity = 0.8)
# Default setting
launch_taxonomic_match_app(min_similarity = 0.7)
# More permissive - allows lower-quality matches
launch_taxonomic_match_app(min_similarity = 0.5)Lower values cast a wider net but may include false positives. Higher values are more conservative but may miss valid matches. The default was raised from 0.3 to 0.7 to reduce spurious suggestions.
Increasing Suggestions
Show more fuzzy match suggestions per name:
# Show top 20 suggestions instead of default 10
launch_taxonomic_match_app(max_suggestions = 20)You can also adjust this interactively in the Review tab using the slider.
Offline Mode
If you do not have a database connection, click “Use offline (cached backbone)” on the login screen. The app:
- Downloads and caches the backbone locally on first use
- Performs string matching entirely in R via
stringdist(trigram-Jaccard) - Supports auto-matching, fuzzy suggestions, and manual search
- Hides the Traits Enrichment tab (requires live connection)
- Displays a “Read-only” badge throughout the session
Function Parameters
launch_taxonomic_match_app(
data = NULL, # Optional: pre-load a data.frame
name_column = NULL, # Optional: pre-select a column name
language = c("fr", "en"),# Interface language (default: "fr")
min_similarity = 0.7, # Fuzzy match threshold (0-1)
max_suggestions = 10, # Max suggestions per unmatched name
mode = "interactive",# Review mode ("interactive" or "batch")
launch.browser = TRUE # Whether to open app in the browser
)Troubleshooting
Connection Issues
Problem: “Failed to connect to database”
Solutions:
# Check connection
db_diagnostic()
# Reset credentials if needed
remove_db_credentials()
setup_db_credentials()Alternatively, use offline mode (click “Use offline (cached backbone)” on the login screen) to work without a live database connection.
No Fuzzy Matches Found
Problem: No suggestions appear for unmatched names
Possible causes: - min_similarity
threshold too high - Taxonomic names contain typos or non-standard
formatting - Names not present in the taxonomic backbone (e.g.,
non-African taxa)
Solutions: - Lower min_similarity:
launch_taxonomic_match_app(min_similarity = 0.5) - Use the
taxonomic level filter to search at genus or family level - Clean input
names (remove extra spaces, fix obvious typos) - Verify names are
African taxa
Slow Matching Performance
Problem: Matching takes very long for large datasets
Solutions: - Enable offline mode:
matching runs locally via stringdist without database
round-trips - Use batch processing instead:
match_taxonomic_names() for programmatic workflows -
Process data in chunks (split large datasets)
When to Use the App vs. Programmatic Approach
Use the Shiny App when:
- Exploring data interactively
- You prefer visual interfaces
- Dataset is small to medium size (<5,000 rows)
- Need to manually review uncertain matches
- Learning the matching process
Use match_taxonomic_names() when:
- Processing large datasets (>5,000 rows)
- Automating workflows in scripts
- Integrating with data pipelines
- Reproducibility is critical (NEVER REMOVE THE COLUMN THAT CONTAINS THE ORIGINAL NAME)
- Batch processing multiple files
Example programmatic approach:
# Load data
my_data <- read.csv("tree_inventory.csv")
# Match names
matched <- match_taxonomic_names(
names = my_data$species_name,
min_similarity = 0.7
)
# Merge back with original data
result <- cbind(my_data, matched)
# Export
write.csv(result, "standardized_inventory.csv", row.names = FALSE)See Also
-
match_taxonomic_names(): Underlying matching function for programmatic use -
query_taxa(): Query taxonomic backbone directly -
match_tax(): Simple taxonomic lookup function -
launch_taxo_backbone_app(): Interactive tool for exploring the taxonomic backbone -
vignette("using-query-plots"): Guide to querying plot data
Tips for Best Results
- Clean your data first: Remove obvious typos, extra whitespace, and special characters
- Understand your data: Know which taxonomic groups are in your dataset
- Use multi-column mode: If you have separate genus/species/family columns, combine them for better matching
- Filter by taxonomic level: Use the level filter in the Review tab to find genus or family matches
- Review match scores: Don’t blindly accept low-similarity matches (<0.6)
- Use checkpoint/resume: The app saves your progress automatically — if you close the browser tab, you can pick up where you left off
-
Document parameters: Note which
min_similarityvalue you used for reproducibility - Cite data sources: Check the Data Sources panel in the Traits tab for citations to include in your methods
Example Workflow
Here’s a complete workflow from start to finish:
# 1. Load your data
trees <- read.csv("forest_inventory.csv")
# Columns: plot_id, tree_number, species_name, dbh, height
# 2. Launch app with data
launch_taxonomic_match_app(
data = trees,
name_column = "species_name",
language = "en",
min_similarity = 0.7
)
# 3. In the app:
# - Authenticate (or choose offline mode)
# - Review automatic matches in the Auto Match tab
# - Use the Review tab to resolve unmatched names
# - Optionally enable WCVP output via the sidebar checkbox
# - Optionally enrich with traits in the Traits Enrichment tab
# (check the Data Sources panel for citations)
# - Export as "forest_inventory_standardized.xlsx"
# 4. Continue analysis with standardized data
standardized <- readxl::read_excel("forest_inventory_standardized.xlsx")
# Now you have clean taxonomic IDs for further analysis!This workflow ensures your taxonomic data is standardized and ready for downstream analyses like diversity metrics, trait-based analyses, or database integration.